Chuanyin DAI, Kai CHEN, Ruiying ZHANG, Xiaojun YANG, Zuohua YIN, Hengjiu TIAN, Zhiming ZHANG, Yan HU, Fumin LEI. 2010: Molecular phylogenetic analysis among species of Paridae, Remizidae and Aegithalos based on mtDNA sequences of COI and cyt b. Avian Research, 1(2): 112-123. DOI: 10.5122/cbirds.2010.0003
Citation:
Chuanyin DAI, Kai CHEN, Ruiying ZHANG, Xiaojun YANG, Zuohua YIN, Hengjiu TIAN, Zhiming ZHANG, Yan HU, Fumin LEI. 2010: Molecular phylogenetic analysis among species of Paridae, Remizidae and Aegithalos based on mtDNA sequences of COI and cyt b. Avian Research, 1(2): 112-123. DOI: 10.5122/cbirds.2010.0003
Chuanyin DAI, Kai CHEN, Ruiying ZHANG, Xiaojun YANG, Zuohua YIN, Hengjiu TIAN, Zhiming ZHANG, Yan HU, Fumin LEI. 2010: Molecular phylogenetic analysis among species of Paridae, Remizidae and Aegithalos based on mtDNA sequences of COI and cyt b. Avian Research, 1(2): 112-123. DOI: 10.5122/cbirds.2010.0003
Citation:
Chuanyin DAI, Kai CHEN, Ruiying ZHANG, Xiaojun YANG, Zuohua YIN, Hengjiu TIAN, Zhiming ZHANG, Yan HU, Fumin LEI. 2010: Molecular phylogenetic analysis among species of Paridae, Remizidae and Aegithalos based on mtDNA sequences of COI and cyt b. Avian Research, 1(2): 112-123. DOI: 10.5122/cbirds.2010.0003
The phylogeny of Paridae and allies has been studied intensively during past decades. However, the phylogenetic relationship among species tends to become increasingly controversial as different genetic markers emerge. In our study, the partial mitochondrial genes cytochrome b (cyt b) and cytochrome c oxidase subunit Ι (COI) were obtained from 15 species that included 10 tits, 4 long-tailed tits and a Chinese penduline tit. Analyses were conducted on the combined cyt b and COI sequences with maximum likelihood and Bayesian algorithms. Based on strong, congruent support among the different temporal partitions and models of sequence evolution, a highly resolved consensus of the relationships among Parids and their allies has been formed. The monophyly of Paridae and Remizidae is strongly supported. However, the monophyly of Paridae and Aegithalos is rejected. This agrees with previous studies using other molecular markers. Our results suggest the promotion of the subgenus Machlolophus from genus Parus to a separate genus. The phylogeny of Aegithalos is robust in the current study. However, by considering differences of both morphological and molecular characters within species, we conclude that more data are needed to define their phylogeny. Based on the patterns of taxonomic diversity and endemism, we suggest the southwestern mountain ranges of China might be the center of origin of the Aegithalos species. Divergence time estimates for the long-tailed tits range from the late Miocene to the Pleistocene (from 5.5 to 0.1 Mya) using a calibration of 2% divergence per million years. In a comparative sense, we found a congruent genetic differentiation among sympatric distribution taxa.
Tits and titmice are small, familiar cavity-nesting songbirds forming the family Paridae which contains 4 genera, 55 to 65 species, depending on different classifications (Harrap and Quinn, 1996; Salzburger et al., 2002; Dickinson, 2003). They are distributed in the entire Holarctic, Oriental and Afrotropical regions with a hot spot of diversity in the Himalayan and Chinese mountain ranges (Päckert et al., 2007). Because of their overall morphological similarity, all but three species have been classified into the genus Parus and the genus is divided into 10 subgenera (Harrap and Quinn, 1996) or 12 species groups (Eck, 1988). The three other genera, Melanochlora, Sylviparus and Pseudopodoces are monotypic. A well-supported, corroborated and presumably accurate phylogeny is essential for an understanding of ecological and behavioral variation within the group. Therefore, in recent years, a plethora of studies using molecular markers, such as allozyme comparisons (Gill et al., 1989), mitochondrial restriction fragment-length polymorphism (RFLP) data (Gill and Slikas, 1992; Gill et al., 1993), DNA-DNA hybridization distances (Sheldon et al., 1992; Slikas et al., 1996) and cytochrome-b sequences (Kvist et al., 1996; Gill et al., 2005; Martens et al., 2006) have been applied to the phylogeny of Paridae. However, the phylogenetic relationship among species tends to become increasingly controversial as different genetic markers emerge. For instance, based on nuclear DNA-DNA hybridization, Parus caeruleus and P. major formed a clade which is the sister group of all other Parids (Sheldon et al., 1992; Slikas et al., 1996), but the phylogeny inferred from cyt b sequences did not support that result (Gill et al., 2005).
Aegithalos, at one time, was classified as a genus in the family Paridae (Gadow, 1883; Hellmayr, 1903; Mayr and Amandon, 1951). However, Snow (1967) considered Aegithalos and a few related genera as a separate family, the Aegithalidae. Phylogenetic analyses based on DNA-DNA hybridization (Sibley and Ahlquist, 1990; Sheldon and Gill, 1996), protein allozyme comparisons (Ohta et al., 2000) and nuclear gene sequences (Barker et al., 2002; Spicer and Dunipace, 2004) suggested that Aegithalos was indeed not Parids and the closest sister group to Parids was the penduline tits, species of Remizidae. However, genera closely related to Aegithalos are still under dispute, as well as the phylogenetic relationships within Aegithalos (Päckert et al., 2010).
In this study, we sampled part of representative taxa of tits, long-tailed tits and penduline tits in an attempt to resolve the relationships among these three groups and also to infer the phylogenetic relationships within tits and long-tailed tits based on two mitochondrial gene sequences, COΙ and cyt b.
Materials and methods
Taxon selection and choice of outgroups
Altogether, twenty-nine individuals of ten species from Paridae, nine individuals of four species from Aegithalos and two Chinese penduline tits were included in this study. Six species had more than one subspecies included in the analysis (Table 1). According to Sibley and Monroe (1990), Sheldon and Gill (1996), Barker et al. (2002), Spicer and Dunipace (2004), an individual of Pica pica, Uragus sibiricus, Garrulax lunulatus and G. ocellatus were selected as outgroups.
Extraction, amplification and sequencing
Genomic DNA was extracted from blood, feathers or tissue specimens using the QIAamp™ DNA Mini Kit as per manufacturer's instructions. Nucleotide sequence data were obtained from the two mitochondrial, cytochrome c oxidase Ⅰ (COI) and cytochrome b (cyt b) genes. PCR amplification and sequencing of cytochrome c oxidase Ⅰ followed the method suggested by Sorenson et al. (1999), while Gill et al. (2005) described protocols for cytochrome b.
For the sequencing reactions, the same primers were used. Both strands of each PCR product were sequenced. For each gene and sample, multiple sequence fragments were obtained by sequencing with different primers. Complete sequences were assembled using Seqman Ⅱ (DNASTAR®). Sequences were compared visually to the original chromatograms to avoid reading errors. Assembled sequences were aligned by eye. All sequences were deposited in GenBank.
Phylogenetic analysis
Based on a priori assumption and partition homogeneity test (p = 0.44), the two mitochondrial genes were analyzed as one data set with a total nucleotide length of 2149 base pairs (bp). The data were analyzed using maximum likelihood (ML, Felsenstein, 1981) and Bayesian inference methods (BI, Rannala and Yang, 1996; Yang and Rannala, 1997; Larget and Simon, 1999). Statistics for nucleotide variation and genetic distance were computed with MEGA 4 (Tamura et al., 2007). A Jukes-Cantor estimate of the number of nucleotide substitutions per site was computed for the cyt b gene (Jukes and Cantor, 1969). Following the suggestion of Nei and Kumar (2000), if the Jukes-Cantor distance is less than 0.05, the p-distance would be used. Otherwise, a more complicated distance model would be employed.
Following alignment, we partitioned the data by genes in order to allow different rates for the various partitions for ML and BI analyses. Nucleotide substitution models were selected separately by genes and then used for different data partitions in reconstruction. However, the TVM + I + G model (−lnL = 15397.6758, K = 9, AIC = 30813.3156) was identified as the best fit for these two genes, using both the likelihood-ratio test (LRT) and Akaike Information Criterion (AIC) implemented in MODELTEST 3.7 (Posada and Crandall, 1998). The parameter values for the model include a symmetric rate matrix specifying relative probabilities for all possible nucleotide changes (Rmatrix = 0.6396 [A-C], 8.0280 [A-G], 1.1611 [A-T], 0.0498 [C-G], 8.0280 [C-T], 1.0000 [G-T]), the proportion of invariant sites (pinvar = 0.6183) and the shape parameter for the gamma distribution of rate variation (shape = 1.2128). The base frequencies were set as follows: A = 0.3263, C = 0.4100, G = 0.1009, T = 0.1628.
ML reconstruction (1000 replicates) was performed in TREEFINDER (Jobb, 2007) and BI performed in MRBAYES 3.1.2 (Ronquist and Huelsenbeck, 2003). In the BI analysis, we ran two analyses of two million generations and trees sampled every 100 generations. One cold and three heated Markov chains were used in our analysis. The trees saved during the "burn-in" phase (the first 200000 generations in our analysis) were discarded. The remaining trees from two runs were used to create a 50% majority rule consensus tree.
Results
Sequence characteristics
We obtained a total of 88 sequences, with their GenBank accession numbers listed in Table 1. No stop codons were identified in a contiguous 1201 bp stretch of the COI gene and 948 bp of cyt b. The overlapping sequences from different PCR products and a single peak in the electropherograms suggest that these sequences do not come from "numts" (nuclear sequences of mitochondrial origin). The average base composition of sequence was skewed, which is similar to that found in previous avian studies (Barhoum and Burns, 2002; Webb and Moore, 2005). The characteristics of these sequences are summarized in Table 2.
Table
1.
List of taxonomic samples and sequences used in the study
Family
Genus
Species and subspecies
Museum No.
Collection locality
GenBank Accession Nos.
cyt b
COΙ
Paridae
Parus
Parus major commixtus
IOZ1254
Guangxi
HM185345
HM185334
Parus major subtibetanus
IOZ5236
Yunnan
HM185346
HM185333
Parus major subtibetanus
IOZ3918
Sichuan
HM185347
HM185332
Parus major artatus
IOZ5661
Jilin
HM185348
HM185331
Parus monticolus yunnanensis
IOZ3633
Sichuan
HM185349
HM185330
Parus monticolus yunnanensis
IOZ2465
Shaanxi
HM185350
HM185329
Parus spilonotus rex
IOZ3026
Fujian
HM185351
HM185328
Parus cyanus
IOZ 781
Xinjiang
HM185352
HM185327
Parus venustulus
IOZ2806
Hubei
HM185353
HM185326
Parus venustulus
IOZ1945
Shaanxi
HM185354
HM185325
Parus ater aemodius
IOZ1094
Gansu
HM185355
HM185324
Parus ater
IOZ2331
Zvolen
HM185356
HM185323
Parus ater ater
IOZ9029
Heilongjiang
HM185357
HM185322
Parus palustris hypermelas
IOZ1239
Gansu
HM185358
HM185321
Parus palustris hypermelas
IOZ7991
Hubei
HM185359
HM185320
Parus palustris brevirostris
IOZ5675
Jilin
HM185360
HM185319
Parus palustris hellmayri
IOZ8680
Shanxi
HM185361
HM185318
Parus palustris hellmayri
IOZ2458
Shaanxi
HM185362
HM185343
Parus montanus baicalensis
IOZ5944
Heilongjiang
HM185363
HM185317
Parus montanus baicalensis
IOZ5945
Heilongjiang
HM185364
HM185316
Parus montanus
IOZ2237
Zvolen
HM185365
HM185315
Parus montanus affinis
IOZ2178
Shaanxi
HM185366
HM185342
Parus dichrous dichrous
IOZ2489
Shaanxi
HM185367
HM185314
Parus dichrous dichrous
IOZ6117
Shaanxi
HM185368
HM185313
Parus cristatus
IOZ2333
Zvolen
HM185369
HM185312
Sylviparus
Sylviparus modestus modestus
KIZglgs1240
Yunnan
HM185370
HM185311
Sylviparus modestus modestus
KIZglgs1241
Yunnan
HM185371
HM185310
Pseudopodoces
Pseudopodoces humilis
IOZ4783
Qinghai
HM185372
HM185309
Pseudopodoces humilis
IOZ4785
Qinghai
HM185373
HM185308
Remizidae
Remiz
Remiz consobrinus
IOZ10640
Liaoning
HM185374
HM185298
Remiz consobrinus
IOZ10652
Liaoning
HM185375
HM185297
Aegithalidae
Aegithalos
Aegithalos caudatus caudatus
IOZ5677
Jilin
HM185376
HM185307
Aegithalos caudatus glaucogularis
IOZ2769
Hubei
HM185377
HM185306
Aegithalos caudatus glaucogularis
IOZ3322
Shaanxi
HM185378
HM185305
Aegithalos concinuus concinnus
IOZ1152
Gansu
HM185379
HM185304
Aegithalos concinuus concinnus
IOZ1499
Hunan
HM185380
HM185303
Aegithalos concinuus concinnus
IOZ3661
Sichuan
HM185381
HM185302
Aegithalos concinuus talifuensis
IOZ5344
Yunnan
HM185382
HM185301
Aegithalos bonvaloti
IOZ3766
Sichuan
HM185383
HM185300
Aegithalos fuliginosus
IOZ2476
Shaanxi
HM185384
HM185337
Timaliidae
Garrulax
Garrulax lunulatus
IOZ2594
Shaanxi
HM185385
HM185341
Garrulax ocellatus
IOZ5083
Yunnan
HM185386
HM185340
Fringillidae
Uragus
Uragus sibiricus
IOZ5870
Heilongjiang
HM185387
HM185344
Corvidae
Pica
Pica pica
IOZ3931
Xinjiang
HM185388
HM185339
Note: IOZ, Institute of Zoology, Chinese Academy of Sciences; KIZ, Kunming Institute of Zoology, Chinese Academy of Sciences.
Because the mean Jukes-Cantor distance was 0.112 ± 0.006 SE (> 0.05) for the cyt b sequence data set, the Tamura-Nei model was chosen to compute the genetic distance (Tamura and Nei, 1993). Sequence divergences within species range from 0% (Parus dichrous) to 2.8% (Aegithalos concinnus), with an average divergence of 1.15%. The mean cyt b sequence distance of Aegithalos was 0.081 ± 0.007 SE, with the lowest 0.002 between A. bonvaloti and A. fuliginosus and the highest 0.118 between A. concinnus and A. caudatus. The mean distance of genus Parus was 0.077 ± 0.005 SE, with the lowest 0.053 between P. major and P. monticolus. Between taxa in Parus and the other two species in the Paridae, average pairwise divergences are as follows: 0.115 ± 0.009 SE to Sylviparus modestus and 0.097 ± 0.007 SE to Pseudopodoces humilis. The distances were 0.16 ± 0.01 SE between Aegithalos and Paridae, 0.18 ± 0.013 SE between Aegithalos and Remizidae, and 0.126 ± 0.01 SE between Paridae and Remizidae.
Phylogenetic relationships
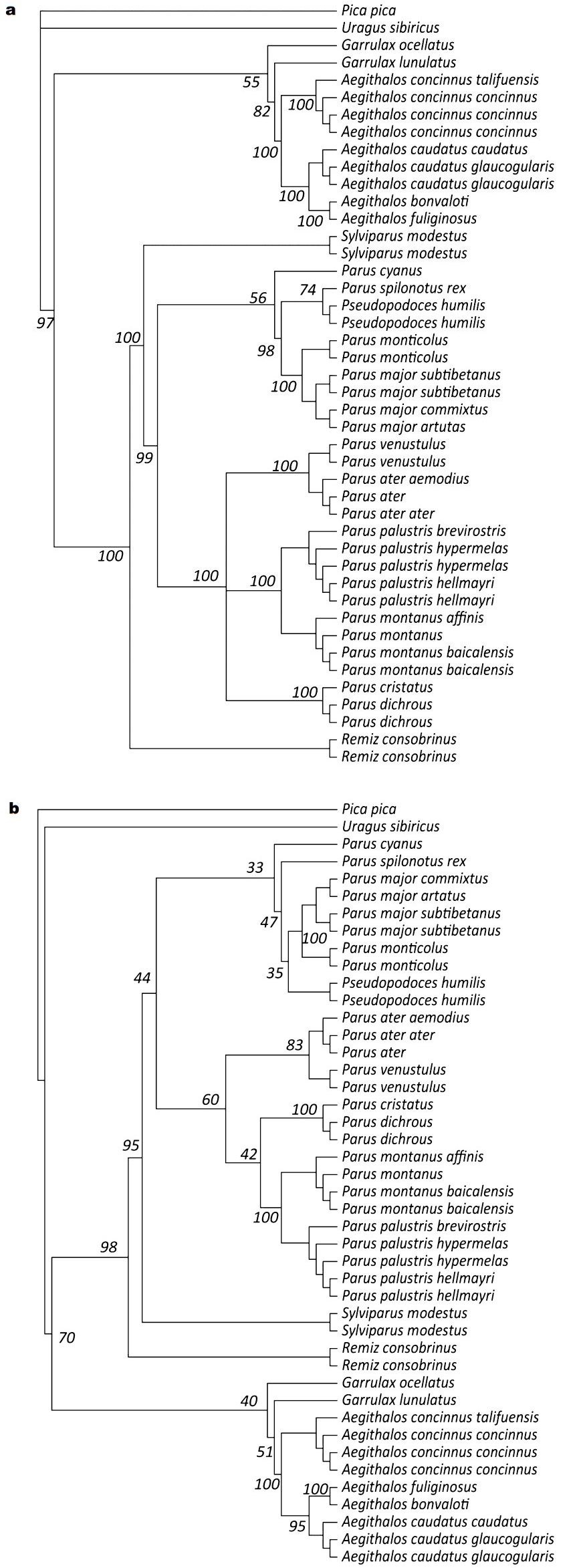
The consensus tree from the Bayesian analysis is identical to the ML tree, except for a few weakly supported nodes. A few nodes that are resolved in the ML tree are polytomies in the Bayesian consensus tree (Fig. 1). In all optimal trees, no species which includes several subspecies were found to be paraphyletic in our study. Paridae, Remizidae, Aegithalos and Garrulax species formed a monophyletic group with high support (bootstrap support = 70%, Bayesian posterior probability = 97%) and branched off to two monophyletic clades. The clade, representing individuals from species of Paridae and Remidae, was highly supported (bootstrap = 98%, BPP = 100%). Remiz consobrinus positions as a sister species to the Paridae, including Sylviparus, Pseudopodoces and the species in Parus.Aegithalos is monophyletic, grouped with two Garrulax species and, with weak support, formed the other clade.
Figure
1.
Trees obtained from the analysis of the combined COΙ and cyt b data: (a) Bayesian and (b) maximum likelihood. The optimal model as estimated by Modeltest following the AIC and LRT was estimated to be TVM + I + G. Support values are indicated to the left of the nodes.
Within Aegithalos, four species are monophyletic and grouped in all optimal trees (Bayesian posterior probability = 100%, bootstrap = 100%). Within the clade, Aegithalos bonvaloti is sister to A. fuliginosus and that pair is sister to A. caudatus. Aegithalos concinnus is sister to these three species. Phylogenetic relationships have strong support (Bayesian posterior probability = 100%, bootstrap > 95%) and are identical in the two phylogenetic trees.
Within the family Paridae, Sylviparus modestus positions as a sister species to the remaining species of Paridae from the two trees. These remaining species form two high-support monophyletic clades in the BI tree (BPP = 99%), but the bootstrap value is rather low (bootstrap = 44%) in the ML tree. One clade includes Parus cyanus, P. monticolus, P. major, P. spilonotus and Ps. humilis. The relationships among these five species are congruent in the two trees except for the position of Ps. humilis. In the two trees, Parus cyanus is a sister species to the remaining species, although the nodal support value is low. Parus major and P. monticolus pair as sister taxa with high support. In the BI tree, P. spilonotus and Ps. humilis pair as sister taxa but in the ML tree, Pseudopodoces humilis is a sister species to P. spilonotus, P. major and P. monticolus. However, the nodal supports are low. Another clade includes P. ater, P. venustulus, P. palustris, P. montanus and two Eurasian crested tits, P. dichous and P. cristatus. Parus palustris and P. montanus, P. ater and P. venustulus, P. dichous and P. cristatus pair as sister taxa with high support. Relationships among these three pairs are unclear because of polytomies in the BI tree and low nodal support value in the ML tree.
Discussion
Phylogenetic relationships among Paridae, Remizidae and Aegithalos
Our data support the classification for the family Paridae proposed by Sibley and Ahlquist (1990). Based on DNA-DNA hybridization, they combined family Remizidae with Paridae, hence the family Paridae includes two subfamilies, Remizinae and Parinae. Sheldon and Gill (1996) studied the phylogeny of song birds using DNA-DNA hybridization and concluded that the sister group of Paridae is the Remizidae. Based on combined myoglobin intron Ⅱ and cytochrome b sequences, Alström et al. (2006) revealed the close relationships between P. major and R. pendulnus. In our study, the Chinese penduline tit (Remizidae) positioned as sister group to the Parids (Paridae) with high support. This may indicate very close relationships between Paridae and Remizidae. Although, in the present study, only one species of Remizidae was tested, the high nodal support (> 95%) signifies that the designated branch is most possibly unaffected by sampling among variation existing in the data (Slikas et al., 1996).
Long-tailed tits, species of Aegithalos, because of their morphological similarity with tits, were earlier believed to be Parids by several authors (Gadow, 1883; Hellmayr, 1903; Mayr and Amandon, 1951). However, Stresemann (1923) described several characteristics of Aegithalos that differ from those of the Parids, such as the presence of a complete juvenile-molt, nest structure and naked hatching. Since then, Aegithalos and two other monotypic or small genera are usually placed in the family Aegithalidae (e.g., Paynter, 1967; Morony et al., 1975; Sibley and Ahlquist, 1990; Sibley and Monroe, 1990). Sturmbauer et al. (1998) suggested a close relationship between Leptopoecile and Aegithalos based on the mitochondrial 16S sequence. Alström et al. (2006) confirmed the close relationship between Aegithalos and Leptopoecile based on the combined myoglobin intron Ⅱ and cytochrome b sequences. Using protein allozyme comparison, Ohta et al. (2000) found the genetic distance between Aegithalos and Parus appears to be at a familial level. In our study, two distinct clades and Aegithalos grouped with two outgroup Garrulax species, in spite of weak nodal support, show distant relationships between Aegithalos and Paridae. In addition, the genetic divergence of cyt b between Aegithalos and Paridae is larger than that between Remizidae and Paridae. Hence, the morphological similarity between tits and long-tailed tits may have resulted from evolutionary convergence.
Phylogenetic relationships within Paridae
Based on plumage pattern, Hellmayr (1903) had recognized eleven subgenera for the genus Parus. However, the delimitation of these subgenera is in dispute. For instance, Wolters (1982) classified the species of Pardaliparus into Pariparus and this classification was supported by molecular analyses (Slikas et al., 1996; Gill et al., 2005). In our study, Parus ater and P. venustulus also formed a closely related group, but the divergence of cyt b is ~7% (uncorrected p-distance). According to Hellmayr's classification, our samples belong to seven subgenera: Cyanistes (P. cyanus), Baeolophus (P. dichous, P. cristatus), Poecile (P. montanus, P. palustrs), Periparus (P. ater), Pardaliparus (P. venustulus), Machlolophus (P. spilonotus) and Parus (P. major, P. monticolus). Similar to previous studies, our data did not reveal phylogenetic relationships among these subgenera. Therefore, the Parus phylogeny needs to be studied further. We intend to add more species and subspecies and new markers, including nuclear loci to classify this challenge in the future.
Although our results came to quite similar conclusions as previous studies, different phylogenetic relationships among species were detected as well. The question of greatest interest is: what is the phylogentically most closely related species to Ps. humilis in Parids? This aberrant and enigmatic Tibetan species, earlier thought to be a corvid, turned out to be a Parid (James et al., 2003). Past phylogenetic analyses based on cyt b gene sequences positioned Ps. humilis as sister species to P. major (James et al., 2003; Gill et al., 2005). However, in optimal trees based on the cyt b and COΙ gene sequences in our study, Ps. humilis is sister to P. spilonotus, although the nodal support is weak. This result may indicate that more data of the species and different markers are needed to determine the phylogenetic status of Parids.
Gill et al. (2005) recommended upgrading six subgenra of Parus into genera for facilitating future evolutionary analyses among Parids. The retained genus Parus only included P. major, P. monticolus, P. xanthogenys, P. spilonotus, P. holsti and the African tits (P. leucomelas, P. niger, P. carpi, P. albiventris, P. leuconotus, P. rufiventris, P. funereus, P. fringillinus, P. fasciiventer, P. thruppi, P. griseiventris, P. cinerascens and P. afer). However, in our present study, we also believe that the genus Machlolophus should be recognized in consideration of the phylogenetic relationships between P. spilonotus and Ps. humilis and distinct plumage pattern between P. major and P. spilonotus (Ohta et al., 2000). Furthermore, in our data, the divergence of the cyt b gene between P. major–P. monticolus group and P. spilonotus (8.4%, uncorrected p-distance) is no less than the divergence among recommended genera (ranging from 7.3% between Pariparus and Baeolophus to 9.7% between Poecile and Machlolophus, uncorrected p-distance).
Intra- and interspecies relationships and history of Aegithalos taxa
On the one hand, intraspecific divergence is significant. The Black-throated Tit (Aegithalos concinnus) includes six highly distinct subspecies. These subspecies are remarkably different in their plumage pattern. Divergence differences of the cyt b gene among these subspecies are also significant. In our data, the pairwise p-distance between specimens from Bengal (India, ssp. iredalei, sequence obtained from GenBank) and Gaoligong (Yunnan, ssp. talifuensis) was 4.5%. p-distance between specimens from Gaoligong and Pingjiang (Hunan, ssp. concinnus) was 6.1%. p-distance between specimens from Bengal and Pingjiang was 5.1%. Eck and Martens (2006) sequenced three specimens, including subspecies ssp. iredalei, ssp. talifuensis and ssp. manipurensis, and they found the pairwise p-distance was 5.1% between ssp. iredalei and ssp. talifuensis, 5.3% between ssp. talifuensis and ssp. manipurensis and 6% between ssp. iredalei and ssp. manipurensis. These four subspecies are reddish-crowned. The other two subspecies A. c. pulchellus and A. c. annamensis are grey-headed. They are obviously different in presence of reddish breast band, the latter of which is absent. Hence, it seems possible that A. concinnus represents an unresolved species swarm (Eck and Martens, 2006).
As for the Long-tailed Tit (Aegithalos caudatus), based on comparative morphology, up to 19 subspecies are currently recognized, divided by Harrap and Quinn (1996) into four groups. Zink et al. (2005) studied the mitochondrial phylogeography, including five subspecies or representatives of A. caudatus group and A. alpinus group of the long-tailed tit. Within their samples, no correspondence between five subspecies and mitochondrial subdivision has been discovered. However, two geographically unsorted lineages were displayed and differentiation seems to exist between the two main groups. In this study, the cyt b divergence of two specimens belonging to the subspecies A. c. caudatus and A. c. glaucogularis, is 1.3%. Thus, phylogeographic structures would be developed if additional subspecies were taken into consideration.
On the other hand, interspecies molecular divergence is small. The morphologically distinct sister species A. fuliginosus and A. bonvaloti are separated by unexpectedly small cyt b divergences (0.2%, uncorrected p-distance) and associated very short branch lengths in the cyt b tree. The pairwise cyt b divergence between the species is comparable to that within populations of the same species in other passerine birds. This could indicate recent separation and divergence from their common ancestor. However, surprisingly, the more slowly evolving nuclear locus β-fibrinogen intron 7 shows relatively greater divergence (2.5%, uncorrected p-distance) than the faster-evolving cyt b (unpublished data). This might be the result of amplification of nuclear pseudogenes instead of mitochondrial DNA (Zhang and Hewitt, 1996; Sorensen and Quinn, 1998). However, our sequences show no evidence of being of nuclear origin. Introgression of mitochondrial DNA seems to be a more likely explanation. A. fuliginosus and A. bonvaloti are not known to hybridize, but their current distributions partly overlap. Hybridization or past hybridization leading to introgression is nevertheless a possibility. Weckstein et al. (2001) argued that introgressive hybridization is the cause of discordant patterns of mitochondrial and allozyme data in the North American sparrows Zonotrichia leucophrys and Z. Atricapilla.
Furthermore, species limits within the A. niveogularis are not, as yet, reliably defined. Harrap and Quinn (1996) proposed the species to be split in two species while Dickinson (2003) proposed a three way split. Therefore, currently, five (Martens and Eck, 1995) or six (Harrap and Quinn, 1996) or seven (Dickinson, 2003) species have been recognized within the genus Aegithalos. Our study includes only a few species and subspecies from Aegithalos because of insufficient or unavailable samples. Recently, Päckert et al. (2010) studied the phylogeny of long-tailed tits and allies based on mitochondrial and nuclear markers and resolved the status of some species on the basis of phylogenetic relationships. However, by considering the long evolutionary time of species and differences within species, both morphological and molecular characters, a study which includes more species and subspecies is necessary to define the phylogeny of Aegithalos.
We suggest that the southwestern mountain ranges of China might be the center of origin of Aegithalos species and that the taxa of this genus colonized new habitats from west to east across China. Supporting this hypothesis are the patterns of taxonomic diversity and endemism. For example, members of all long-tailed tits are found in the areas of southwestern China except A. niveogularis. The oldest linage in the genus in our study is the A. concinnus talifuensis which is distributed in Yunnan Province of China. The historical biogeography can be deduced by the relationships among species and the areas of species distribution (Gill et al., 2005). For instance, the closely related phylogenetic relationships between Aegithalos fuliginosus and A. bonvaloti is accompanied by a partly overlapped distribution. Aegithalos caudatuscaudatus is older than A. c. glaucogularis in our phylogenetic analysis and is thus distributed farther from the original areas than A. c. glaucogularis. Given a rough calibration of 2% divergence per million years, we hypothesize that the separation of A. concinnus may occur ~5.5 Mya, on the basis of the estimate of 11% sequence divergence between A. concinnus and other three species. Then, about 4.5 Mya ago, A. caudatus was divided and colonized the palearctic region. More recently, separation between Aegithalos fuliginosus and A. bonvaloti is hypothesized to have taken place in the late Pleistocene, i.e., ~100000 years ago. However, that break may be earlier than late Pleistocene, for the possible introgression between two species may obscure the real separation time.
Congruent genetic differentiation among co-distributed species
Comparative phylogeography helped elucidate the relative effect of shared historical earth events on current patterns of biodiversity by comparing historical patterns of gene flow and divergence among species that overlap in time and space (Hickerson et al., 2010). In other words, co-distribution species may have similar population genetic differentiation or congruent phylogeographical patterns as a result of sharing common environmental and geological changes. In our study, the Great Tit (Parus major) and the Black-throated Tit (A. concinnus) overlap in southern and southwestern China, the Coat Tit (P. ater), the Willow Tit (P. montanus), the Marsh Tit (P. palustris) and the Long-tailed Tit are distributed in the northeast, center and west of China. Although our sample size was small, we found congruent genetic differentiation among these sympatric distribution taxa. The great tit and the black-throated tit have similar phylogeographical patterns. They differentiated into two clades, one including the samples of Yunnan Province and the other one belonging to the other sample locations in China. Similarly, the coat tit, the marsh tit, the willow tit and the long-tailed tit also divided into two branches, one covering the areas of northeastern China and Europe and the other including the species distribution in central and western China. These phylogeographical patterns or population structures of sympatric species may indicate that the historical earth events they experienced, including climatic and geological, had almost the same effect on these closely related taxa.
Acknowledgements
We are grateful to Tao Li for laboratory assistance and anonymous reviewers who provided helpful comments to the improvement of the manuscript. This work was financially supported by the National Natural Science Foundation of China (Grant No. 30870270) and the National Science Funds for Distinguished Young Scientists (No. 30925008) to Fumin Lei.
Alström P, Ericson PGP, Olsson U, Sundberg P. 2006. Phylogeny and classification of the avian superfamily Sylvioidea. Mol Phylogenet Evol, 38:381–397.
Barker FK, Barrowclough GF, Groth JG. 2002. A phylogenetic hypothesis for passerine birds: taxonomic and biogeographic implications of an analysis of nuclear DNA sequence data. Proc Roy Soc London B, 269:295–308.
Barhoum DN, Burns KJ. 2002. Phylogenetic relationships of the wrentit based on mitochondrial cytochrome b sequences. Condor, 104:740–749.
Dickinson EC. 2003. The Howard and Moore Complete Checklist of the Birds of the World. Princeton University Press, Princeton, New Jersey.
Eck S. 1988. Gesichtspunkte zur Art-Systematik der Meisen (Paridae). Zool Abh Mus Tierkd Dresden, 43:101–134. (in German)
Eck S, Martens J. 2006. Systematic notes on Asian birds. 49. A preliminary review of the Aegithalidae, Remizidae and Paridae. Zool Med Leiden, 80-5(1):1–63.
Felsenstein J. 1981. A likelihood approach to character weighting and what it tells us about parsimony and compatibility. Biol J Linn Soc, 16:183–196.
Gadow H. 1883. Catalogue of the Passeriformes, or Perching Birds, in the Collection of the British Museum. British Museum (Nat. Hist.), London.
Gill FB, Funk DH, Silverin B. 1989. Protein relationships among titmice (Parus). Wilson Bull, 101(2):182–197.
Gill FB, Slikas B. 1992. Patterns of mitochondrial DNA divergence in North American crested titmice. Condor, 94:20–28.
Gill FB, Mostrom A, Mack AL. 1993. Speciation in North American chickadees: Ⅰ. Patterns of mtDNA genetic divergence. Evolution, 47:193–212.
Gill FB, Slkas B, Sheldon F H. 2005. Phylogeny of titmice (Paridae): Ⅱ. Species relationships based on sequences of the mitochondrial cytochrome-b gene. Auk, 122:121–143.
Harrap S, Quinn D. 1996. Nuthatches, Creepers and Titmice. Princeton University Press, Princeton, New Jersey.
Hellmayr CE. 1903. Paridae, Sittidae und Certhiidae. Das Tierreich. 18:1–255. R. Friedländer und Sohn, Berlin. (in German)
Hickerson MJ, Carstens BC, Cavender-Bares J, Crandall KA, Graham CH, Johnson JB, Rissler L, Victoriano PF, Yoder AD. 2010. Phylogeography's past, present, and future: 10 years after Avise, 2000. Mol Phylogenet Evol, 54:291–301.
James HF, Ericson PGP, Slikas B, Lei F, Gill FB, Olson SL. 2003. Pseudopodoces humilis: a misclassified terrestrial tit (Aves: Paridae) of the Tibetan Plateau: Evolutionary consequences of shifting adaptive zones. Ibis, 145:185–202.
Jobb G. 2007. TREEFINDER Version of November 2007. Munich, Germany. .
Jukes TH, Cantor CR. 1969. Evolution of protein molecules. In: Munro RE (ed) Mammalian Protein Metabolism. Academic Press, New York. pp. 21–132.
Kvist L, Ruokonen M, Orell M, Lumme J. 1996. Evolutionary patterns and phylogeny of tits and chickadees (genus: Parus) based on the sequence of the mitochondrial cytochrome b gene. Ornis Fenn, 73:145–156.
Larget B, Simon DL. 1999. Markov chain Monte Carlo algorithms for the Bayesian analysis of phylogenetic trees. Mol Biol Evol, 16:750–759.
Martens J, Eck S. 1995. Towards an ornithology of the Himalayas: Systematics, Ecology and Vocalizations of Nepal Birds. Bonner Zool Monogr, 38:1–445.
Martens J, Tietze DT, Sun YH. 2006. Molecular phylogeny of Parus (Periparus), a Eurasian radiation of tits (Aves: Passeriformes: Paridae). Zool Abh Mus Tierkd Dresden, 55:103–120.
Mayr E, Amandon D. 1951. A classification of recent birds. Am Mus Novit, 1496:1–42.
Morony JJ Jr, Bock WJ, Farrand Jr J. 1975. Reference List of the Birds of the World. American Museum of Natural History, New York.
Nei M, Kumar S. 2000. Molecular Evolution and Phylogenetics. Oxford University Press, New York.
Ohta N, Kusuhara S, Kakizawa R. 2000. A study on genetic differentiation and phylogenetic relationships among East Asian titmice (Family: Paridae) and relatives. Jpn J Ornithol, 48:205–218.
Paynter RA. 1967. Checklist of Birds of the World. Cambridge, Mass.
Päckert M, Martens J, Tietze DT, Dietze C, Wink M, Kvist L. 2007. Calibration of a molecular clock in tits (Paridae)—nucleotide substitution rates of mitochondrial genes deviate from the 2% rule. Mol Phylogenet Evol, 44:1–14.
Päckert M, Martens J, Sun Y. 2010. Phylogeny of long-tailed tits and allies inferred from mitochondrial and nuclear markers (Aves: Passeriformes, Aegithalidae). Mol Phylogenet Evol, doi: .
Posada D, Crandall KA. 1998. MODELTEST: testing the model of DNA substitution. Bioinformatics, 14:817–818.
Rannala B, Yang Z. 1996. Probability distribution of molecular evolutionary trees: a new method of phylogenetic inference. J Mol Evol, 43:304–311.
Salzburger W, Martens J, Nazarenko AA, Sun YH, Dallinger R, Sturmbauer C. 2002. Phylogeography of the Eurasian Willow Tit (Parus montanus) based on DNA sequences of the mitochondrial cytochrome-b gene. Mol Phylogenet Evol, 24:26–34.
Sheldon FH, Slikas B, Kinnarney M, GillF B, Zhao E, Silverin B. 1992. DNA-DNA hybridization evidence of phylogenetic evidence relationships among major lineages of Parus. Auk, 109:173–185.
Sheldon FH, Gill FB. 1996. A reconsideration of songbird phylogeny, with emphasis on the evolution of titmice and their sylvioid relatives. Syst Biol, 45:473–495.
Sibley CG, Ahlquist JE. 1990. Phylogeny and Classification of Birds: A Study in Molecular Evolution. Yale University Press, New Haven.
Sibley CG, Monroe BL Jr. 1990. Distribution and Taxonomy of Birds of the World. Yale University Press, New Haven.
Slikas B, Sheldon FH, Gill FB. 1996. Phylogeny of titmice (Paridae): Estimate of relationships among subgenera based on DNA-DNA hybridization. J Avian Biol, 27:70–82.
Snow DW. 1967. The families Aegithalidae, Remizidae and Paridae. pp 52–124. In: Paynter RA Jr (ed) Check-list of Birds of the World. A Continuation of the Work of James L. Peters. Cambridge, Mass. Ⅻ: pp ⅰ–ⅸ, 1–495.
Sorenson MD, Ast JC, Dimcheff DE, Yuri T, Mindell D. 1999. Primers for a PCR-based approach to mitochondrial genome sequencing in birds and other vertebrates, Mol Phyl Evol, 12:105–114.
Sorensen MD, Quinn TW. 1998. Numts: a challenge for avian systematics and population biology. Auk, 115:214–221.
Spicer GS, Dunipace L. 2004. Molecular phylogeny of songbirds (Passeriformes) inferred from mitochondrial 16S ribosomal RNA gene sequences. Mol Phylogenet Evol, 30:325–335.
Sturmbauer C, Berger B, Dallinger R, Föger M. 1998. Mitochondrial phylogeny of the genus Regulus and implications on the evolution of breeding behavior in sylvioid songbirds. Mol Phylogenet Evol, 10:144–149.
Stresemann E. 1923. Ueber die systematische Stellung der Paradoxornithinae. Verhandl Ornithol Gesells Bayern, 15:387–390. (in German)
Tamura K, Nei M. 1993. Estimation of the number of nucleotide substitutions in the control region of mitochondrial-DNA in humans and chimpanzees. Mol Biol Evol, 10:512–526.
Tamura K, Dudley J, Nei M, Kumar S. 2007. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol, 24:1596–1599.
Webb DM, Moore WS. 2005. A phylogenetic analysis of woodpeckers and their allies using 12S, Cyt b, and COI nucleotide sequences (class Aves; order Piciformes). Mol Phylogenet Evol, 36:233–248.
Weckstein JD, Zink RM, Blackwell-Rago RC, Nelson DA. 2001. Anomalous variation in mitochondrial genomes of White-crowned (Zonotrichia leucophrys) and Golden-crowned (Z. atricapilla) Sparrows: pseudogenes, hybridization, or incomplete lineage sorting? Auk, 118:231–236.
Wolters HE. 1982. Die Vogelarten der Erde. Paul Parey, Hamburg and Berlin.
Yang Z, Rannala B. 1997. Bayesian phylogenetic inference using DNA sequences: a Markov chain Monte Carlo method. Mol Biol Evol, 14:717–724.
Zhang D, Hewitt GM. 1996. Nuclear integrations: challenges for mitochondrial DNA markers, Trends Ecol Evol, 11:247–251.
Zink RM, Pavlova A, Drovetski S, Rohwer S. 2008. Mitochondrial phylogeographies of five widespread Eurasian bird species. J Ornithol, 149:399–413.

 DownLoad:
DownLoad:





 Email Alerts
Email Alerts RSS Feeds
RSS Feeds